
Fewer MedTech start-ups are coming to market in the EU – but would the newly revised MDR/IVDR rules be able to shake things up?
Whilst the EU has continued to produce new MedTech startups over the last two - three years, the pace of new companies coming to market has slowed down compared with the pre MDR and COVID years and growth is concentrated in a few large countries with strong funding and regulatory support. The proposed MDR/IVDR revisions announced in December 2025 and AI Act alignment are hoped to reverse this trend from 2026 onward and instil more agility in the EU MedTech ecosystem.
HEALTHCARE REGULATIONHEALTHCARE INNOVATION STRATEGYARTICLES
Irene Petre
3/5/20265 min read
Whilst the EU has continued to produce new MedTech startups over the last 2–3 years, the pace of new companies coming to market has slowed down compared with the pre‑MDR and COVID years and growth is concentrated in a few large countries with strong funding and regulatory support such as Germany, France, Ireland, Spain, Italy.
SMEs and startups still represent c. 95% of the European MedTech industry, but fewer are reaching late clinical or CE‑marking stages compared with 2018–2020. The bottleneck is not company formation, but market entry and certification.
The EU remains one of the world’s largest MedTech ecosystems with over 30k start-ups and SMEs, but notes slower company scaling and longer time‑to‑market since MDR/IVDR implementation. There is more focus on Digital health and AI‑enabled tools, incremental innovation and Platform technologies with staged regulatory strategies. More startups are delaying CE marking or launching first outside the EU in places like Switzerland, the UK and the US.
Funding data (which is often the best proxy for startup formation and progression) in the EU shows a slight decline between 2022-24 – which reflects macroeconimic tightening rather than a collapse in innovation but it is of course a factor impacting on the number of startups able to progress to certification (https://sifted.eu/scout/medtech-q1-2024?utm_source=copilot.com):
Several structural factors explain why startup numbers have not accelerated
These factors include:
MDR/IVDR certification costs and timelines disproportionately affect startups compared to larger more established organisations that have more money, expertise and resources to deal with regulation
Notified Body (NB) capacity constraints impact the timelines in which cases can be progressed and certifications issued, delaying market entry for start-ups
Investor caution around regulatory risk has been on the rise in the last 2 – 3 years
There was a shift towards capital‑efficient digital and AI solutions with stagedregulatory exposure.
The EU Medical Device Regulation (EU MDR) was introduced to improve patient safety standards, strengthen clinical evidence and improve product lifecycle oversight. Yet several years after its implementation, many medtech manufacturers (not just start-ups but also well established businesses) continue to struggle with the amount of paperwork and (sometimes duplicated) information required, compliance bottlnecks, certification delays, and soaring budgets.
But EU innovation has not declined, only that commercialisation rate has slowed down (fewer startups get to commercialisation), time to reach the market has increased and the process is more selective as only well prepared start-up can get to the finish line in a reasonable amount of time.(https://www.medtecheurope.org/resource-library/medtech-europes-facts-figures-2024/?utm_source=copilot.com). Others can also make it but with extensive costs and delays.
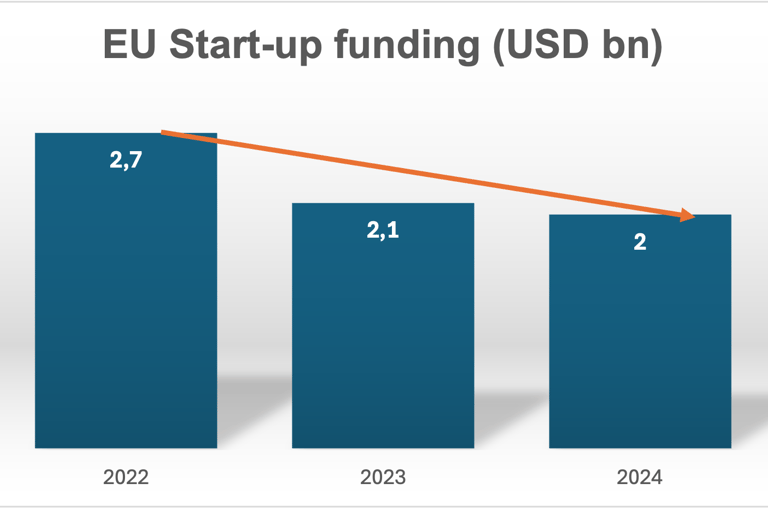
CAGR -13,9%
Start-ups that succeed tend to initially target lower risk classes and delay CE marking registration
Start-ups that are successful tend to:
Design for lower initial risk classes
Build AI lifecycle, Post-Market Surveillance (PMS) readiness and Post-Market Clinical Follow-up (PMCF) integration early on
Delay full CE marking until product‑market fit is clearer
Set measurable objectives from the beginning
Bring meaningful real-world validated clinical evidence
Have strong alignment with State of the Art processes and cybersecurity
Have clear consistent intended use statements across all channels and materials
Performs a strong benefit - risk analysis
Treat the MDR as a continuous lifecycle obligation – meaning these successful startups see the MDR as a mandatory process that integrates clinical evaluation and real-world data (RWD), risk management, PMS and regulatory planning from product development phases all the way through post-market launch monitoring.
Many EU based startups now incorporate or pilot in the UK or Switzerland and then return to the EU later to obtain the CE marking. That is because UK and Swiss ecosystems currently offer faster regulatory learning, better investor confidence and earlier clinical deployment.


MDR_IVDR revisions are being drafted to cut bureaucracy and help more start-up register and come to market
The proposed MDR/IVDR revisions and AI Act alignment are expected to improve this picture from 2026 onward, especially for software‑heavy startups (SaMD).
Some of the key proposed changes that are likely to make a difference for Health Tech start-ups include:
some classification rules are being adjusted and relaxed so that certain device categories will result in lower risk classes, including reusable surgical instruments, accessories to active implantable devices and software
to better distinguish software with life threatening output (so which may directly cause death or irreversible deterioration of health), which likely will remain in higher classes from software with more limited impact (particularly where the digital tool's contribution to clinical decision‑making is indirect or where impact on patient outcomes is limited), These types of SaMD may be down‑classified from class 2a or in some cases from class 2b to class 1 in the future
make the MDR certification more affordable, particularly for SMEs and start-ups - annual monitoring with joint assessment teams is likely to replace the need for recertification every five years, while structured dialogue formalises manufacturer-notified body interaction throughout conformity assessment
representative device sampling, risk-based surveillance intervals and remote audit options are trying to reduce assessment workload for Notified Bodies without compromising independent oversight - thus making the certification process more predictable, affordable, transparent and simple
·startups will be able to submit Technical documentation, EU declarations of conformity and assessment reports in digital format, making the whole process faster and bringing costs down for startups (New article 52b of the MDR and article 48b of the IVDR)
more product information will be provided digitally, particularly for professional‑use devices (article 110 of the MDR, 103a for the IVDR) and in environments where internet access and electronic documentation systems are reliable. This can allow manufacturers to update safety information more dynamically
maintaining accurate, up-to-date data in Eudamed or linked systems could become increasingly important as authorities may rely more heavily on these systems for market surveillance purposes
· For online and digitally enabled medtech and diagnostic businesses, the potential convergence of UDI, digital labelling, cyber incident reporting and Eudamed registration suggests that regulatory compliance considerations may extend beyond technical and quality documentation into digital product design and commercial operations.
proposed reforms position MDR and IVDR as the primary framework for medical AI, reducing risk of duplicative conformity assessments under AI Act
The new proposed obligation to report actively exploited vulnerabilities and severe cyber incidents within 30 days to both regulatory and cyber authorities is an important potential change, as it would require significant organisational readiness – start-ups will need to enhance their monitoring and incident management capabilities.
These are not exhaustive - there are potential changes. Overall one key takeaway is that the proposed MDR/IVDR revisions introduce targeted clarifications for medical device software (MDSW) and AI‑based devices, especially around classification (Rule 11), lifecycle expectations, PMS, and risk management, and these changes will materially affect how startups prepare for NB review. The revisions aim to make the framework more proportionate and innovation‑friendly, but they also raise concerns about reduced scrutiny and inconsistent definitions, which could create uncertainty for early‑stage companies.


IGEA Healthcare
Strategic Advisory for Life Sciences
Switzerland, UK, Italy
contact@igeahealthcare.com
© 2025. All rights reserved.